
Deep Learning models for interpreting biological data with prior knowledge
Cancer subtype classification and modeling by pathway attention and propagation
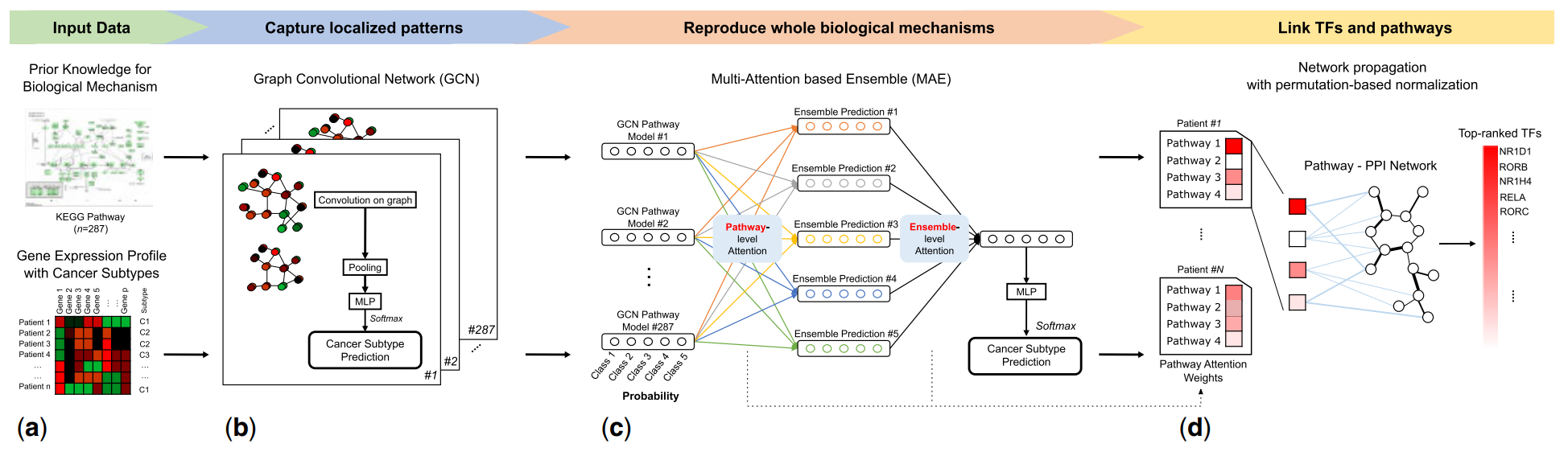
- Designing a pathway-based explainable deep learning model by graph convolutional network and attention mechanism
- Predicting cancer subtypes using gene expression data and pathway information
- Paper accepted by Bioinformatics journal
Learning Cell-Type-Specific Gene Regulation Mechanisms by Multi-Attention Based Deep Learning with Regulatory Latent Space
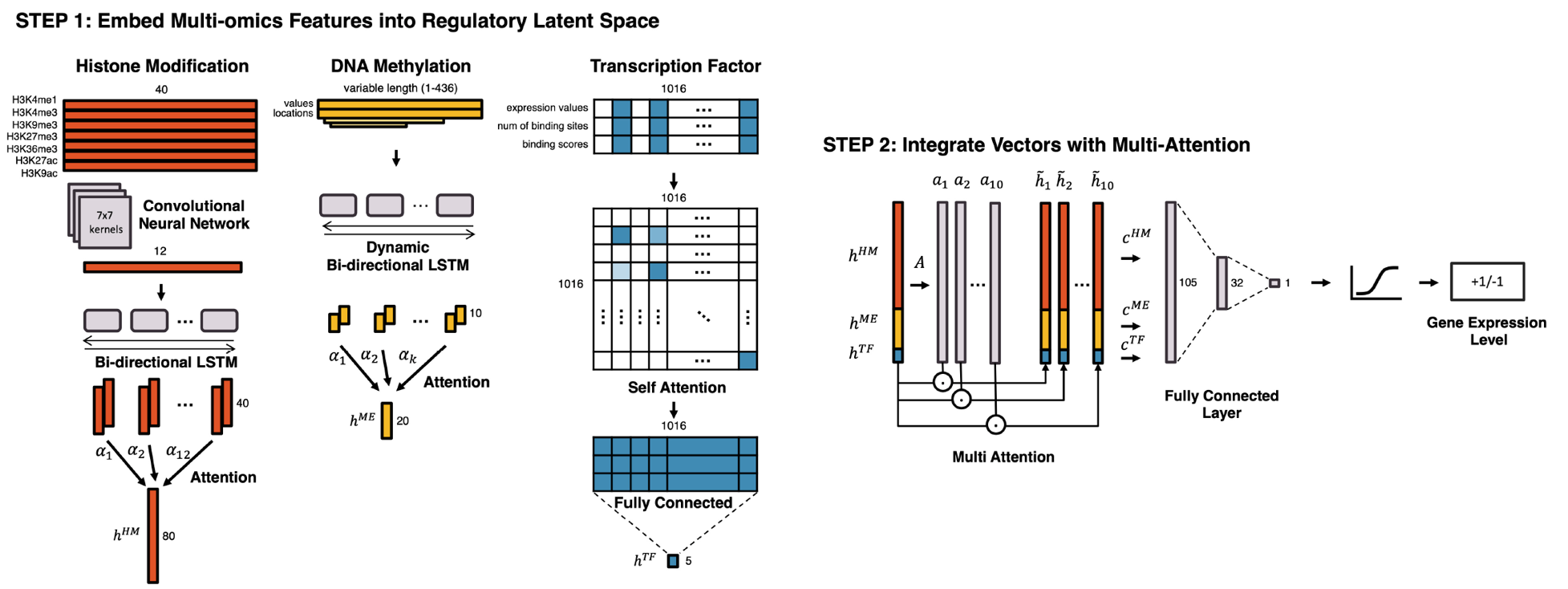
- Designing multi-modal deep learning model for learning gene regulation mechanism
- Elucidating epigenetic gene regulation mechanism by various attention layers
- Paper accepted by Frontiers in Genetics journal
Multi-layered Knowledge Graph Neural Network Reveals Pathway-level Agreement of Three Breast Cancer Multi-gene Assays
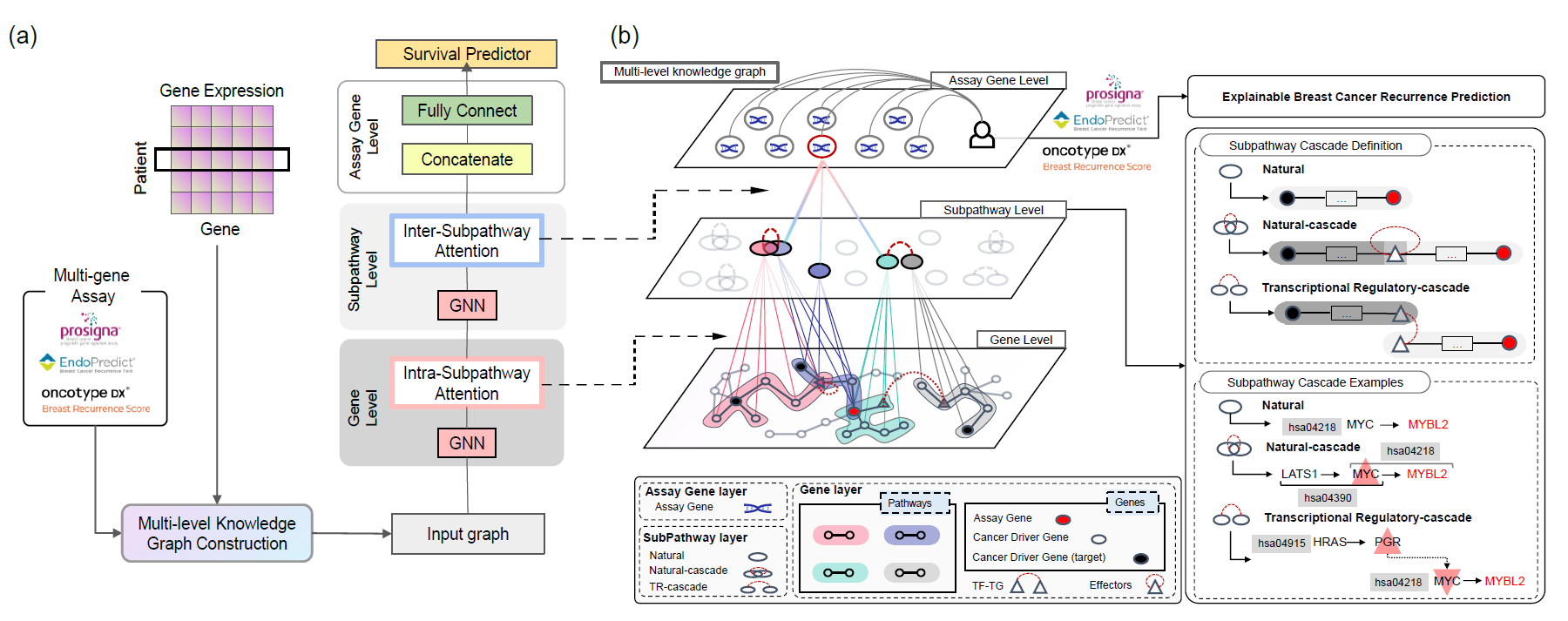
- Designing an explainable deep learning model by integrating intra- and inter- pathway level attention
- Identification of shared regulatory mechanisms of three breast cancer multi-gene assays
- Under review in SCIE journal
AI with graph structured data in biomedical domains
Sparse Structure Learning via Graph Neural Networks for Inductive Document Classification
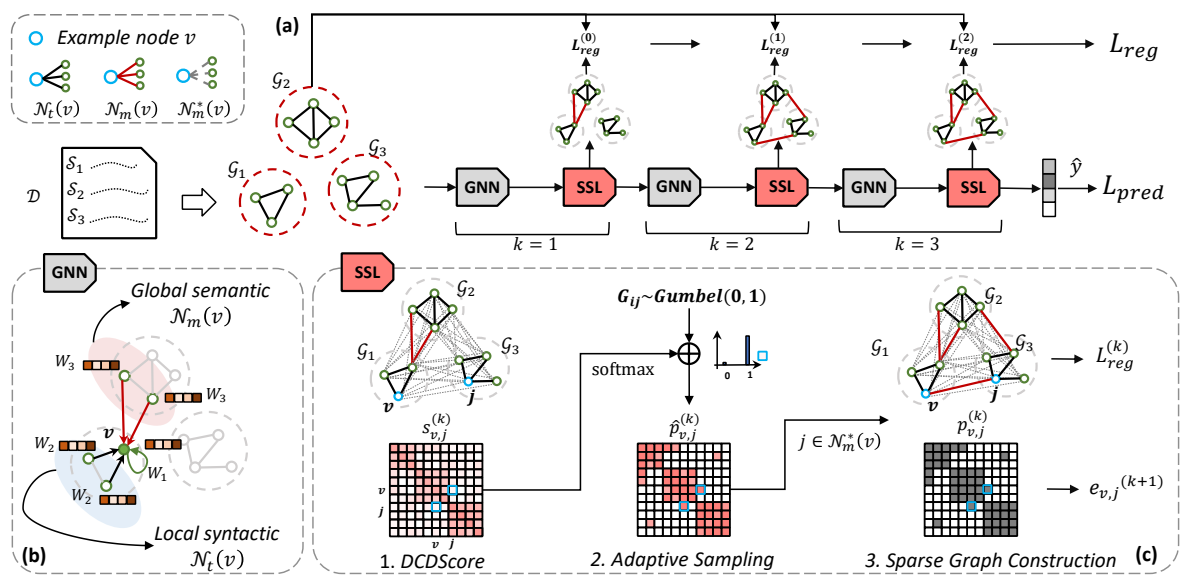
- A novel GNN-based sparse structure learning model for inductive document classification
- Employing structure learning to sparsely select edges between words by considering dynamic contextual dependencies
- Paper accepted by AAAI 2022
Biomedical knowledge graph learning for drug repurposing by extending guilt-by-association to multiple layers
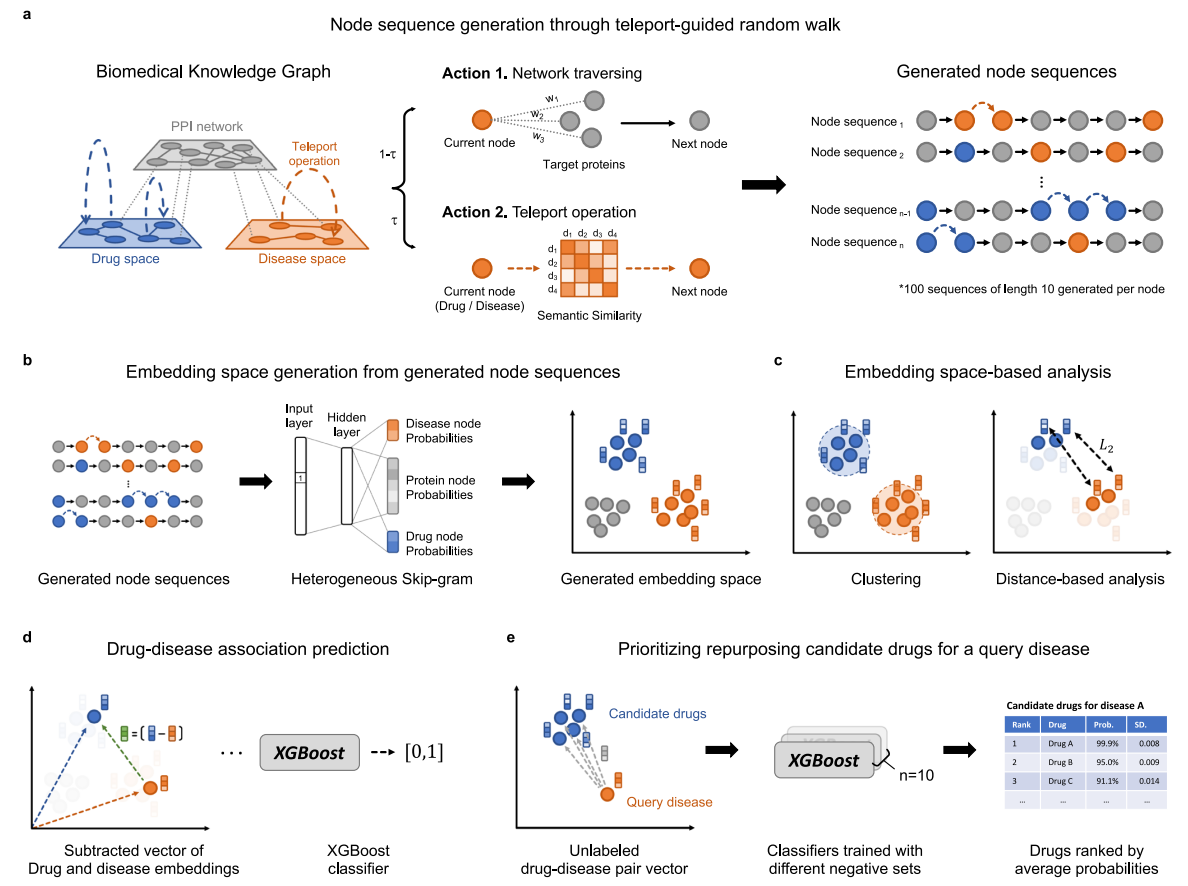
- A semantic multi-layer guilt-by-association approach that leverages the principle of guilt-by-association - “similar genes share similar functions” at the drug-gene-disease level
- Designing a semantic information-guided random walk to generate embeddings of drugs and disease in a unified embedding space
- Paper accepted by Nature Communications journal
Cheminformatics and AI in Drug discovery
Multi-Task Informed Learnable Prototypes on Few Shot learning for Molecular Property Prediction
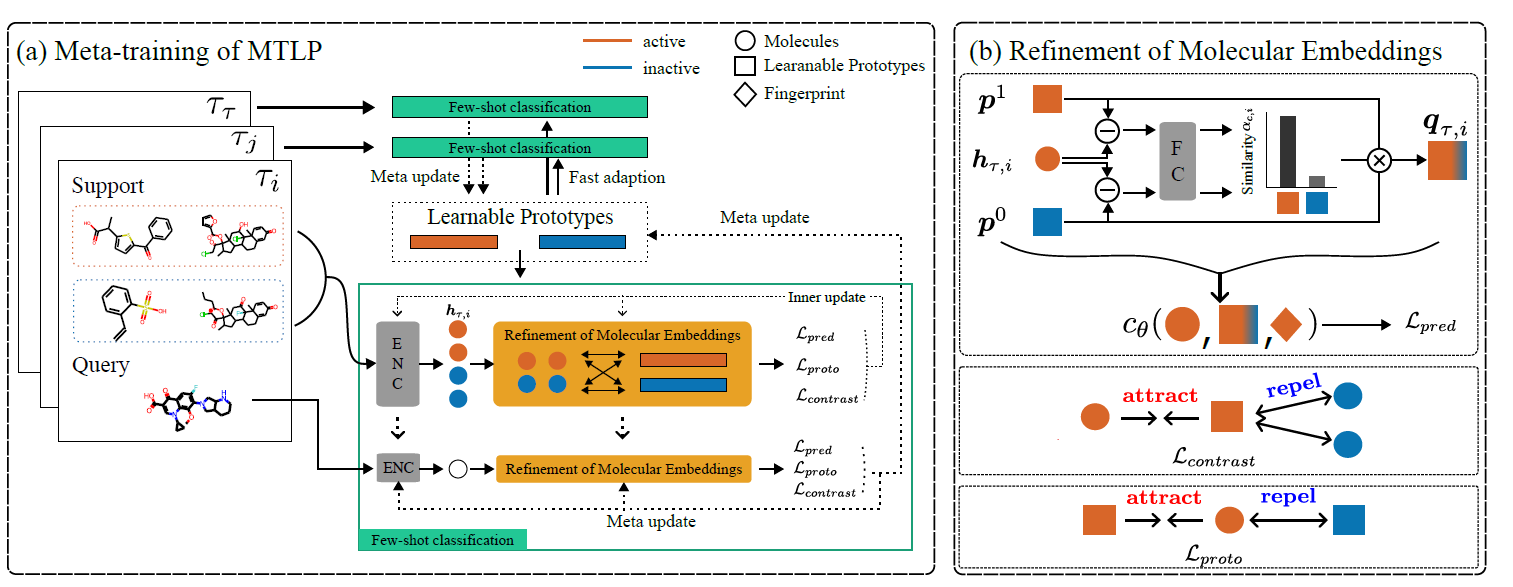
- Leveraging shared knowledge across multiple molecular properties in few-shot learning
- Incorporating a stochastic attention mechanisms to reflect information from multiple assays
- Under review in International Conference
Dual Representation Learning for Predicting Drug-side Effect Frequency using Protein Target Information
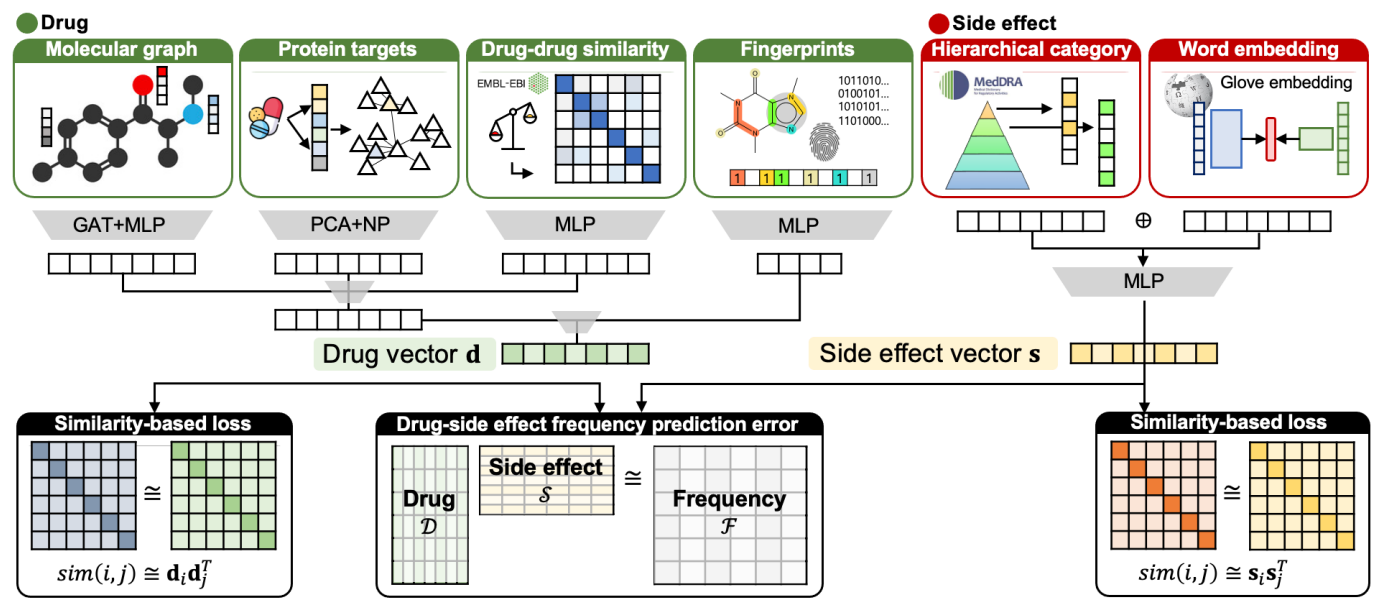
- Utilizing heterogenous features of drugs and side effects to get improved latent representations
- Compensating for the drugs without clear target proteins using the Adaboost method
- Paper accepted by IEEE Journal of Biomedical and Health Informatics journal
Improved drug response prediction by drug target data integration via network-based profiling
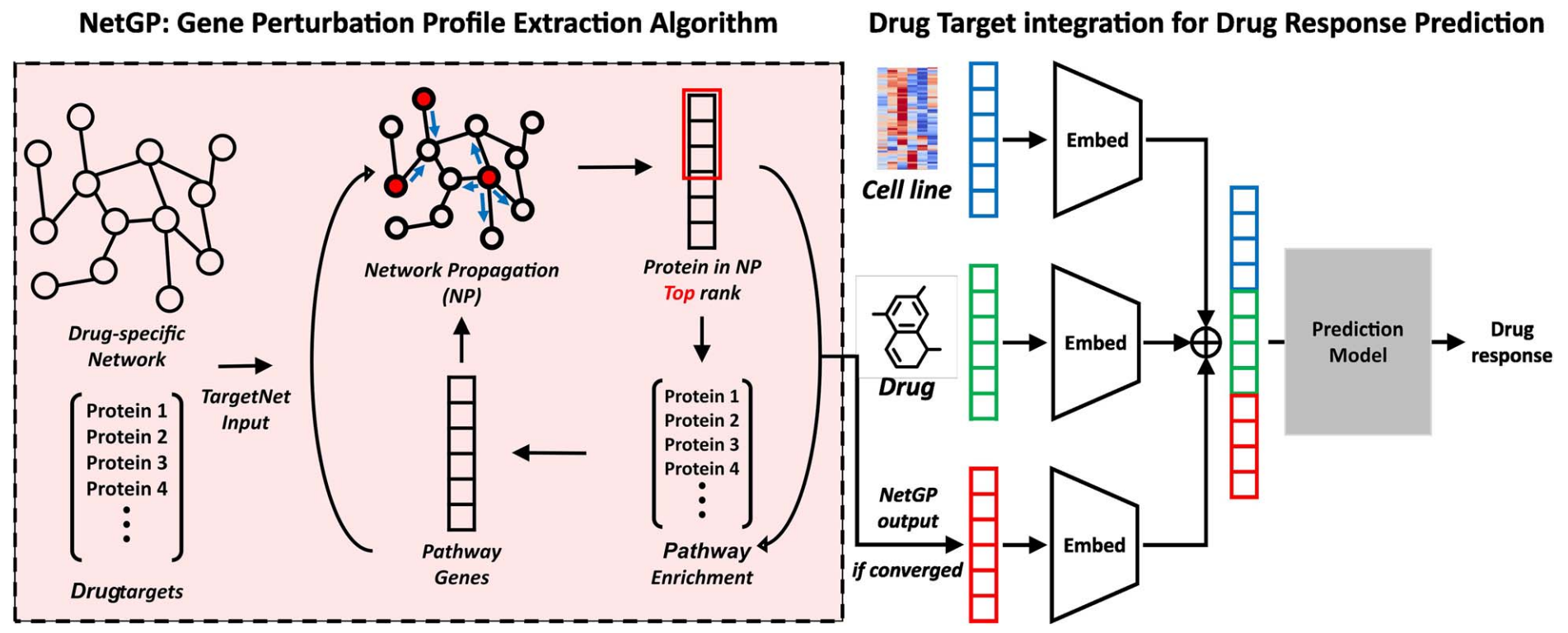
- A novel model-agnostic framework to enhance performances of existing deep learning models for drug response prediction
- Network-based estimation of gene perturbation by drug treatment
- Paper accepted by Briefings in Bioinformatics journal